Article Text

Abstract
Pulmonary hypertension is a progressive and often fatal cardiopulmonary condition characterised by increased pulmonary arterial pressure, structural changes in the pulmonary circulation, and the formation of vaso-occlusive lesions. These changes lead to increased right ventricular afterload, which often progresses to maladaptive right ventricular remodelling and eventually death. Pulmonary arterial hypertension represents one of the most severe and best studied types of pulmonary hypertension and is consistently targeted by drug treatments. The underlying molecular pathogenesis of pulmonary hypertension is a complex and multifactorial process, but can be characterised by several hallmarks: inflammation, impaired angiogenesis, metabolic alterations, genetic or epigenetic abnormalities, influence of sex and sex hormones, and abnormalities in the right ventricle. Current treatments for pulmonary arterial hypertension and some other types of pulmonary hypertension target pathways involved in the control of pulmonary vascular tone and proliferation; however, these treatments have limited efficacy on patient outcomes. This review describes key features of pulmonary hypertension, discusses current and emerging therapeutic interventions, and points to future directions for research and patient care. Because most progress in the specialty has been made in pulmonary arterial hypertension, this review focuses on this type of pulmonary hypertension. The review highlights key pathophysiological concepts and emerging therapeutic directions, targeting inflammation, cellular metabolism, genetics and epigenetics, sex hormone signalling, bone morphogenetic protein signalling, and inhibition of tyrosine kinase receptors.
- Pulmonary disease, chronic obstructive
- Vascular diseases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Definition, classification, and epidemiology of pulmonary hypertension
Pulmonary hypertension covers a group of cardiopulmonary diseases defined at the Sixth World Symposium for Pulmonary Hypertension as a mean pulmonary artery pressure of >20 mm Hg, pulmonary artery occlusion pressure ≤15 mm Hg, and pulmonary vascular resistance >3 Woods units (1 Woods unit=80 dynes × sec/cm5).1 Pulmonary hypertension is clinically divided into five groups: pulmonary arterial hypertension (group 1), pulmonary hypertension caused by left heart disease (group 2), pulmonary hypertension caused by lung diseases, hypoxia, or both (group 3), chronic thromboembolic pulmonary hypertension and pulmonary hypertension caused by pulmonary artery obstructions (group 4), and pulmonary hypertension caused by unclear or multifactorial mechanisms (group 5).2 Pulmonary hypertension caused by left heart disease (group 2) followed by pulmonary hypertension caused by chronic lung disease (group 3) comprise the largest population of patients with pulmonary hypertension worldwide (table 1).3
Pulmonary hypertension is characterised by progressive pulmonary vascular remodelling and, if left untreated, leads to right ventricular failure and death.4–6 Estimates of 1% of the global population and up to 10% of individuals aged >65 years with pulmonary hypertension have been reported.3 Many common conditions and diseases are complicated by pulmonary hypertension or right ventricular failure, or both, including HIV infection, chronic liver disease, connective tissue disease, congenital heart disease, schistosomiasis, heart failure with reduced ejection fraction, heart failure with preserved ejection fraction, valvular heart disease, chronic obstructive pulmonary disease, pulmonary fibrosis, sleep disordered breathing, pulmonary emboli, myeloproliferative disorders, chronic haemolysis, and end stage renal disease (table 1).4 7–10 The presence of pulmonary hypertension in all of these conditions is associated with worse outcomes.9 10
Pulmonary hypertension: classification, epidemiology, haemodynamic characteristics, and treatments
Among the five pulmonary hypertension groups, pulmonary arterial hypertension (group 1) is one of the most aggressive types of pulmonary hypertension.11 Because most of the progress in the specialty has been made in the research and clinical care of pulmonary arterial hypertension, this type of pulmonary hypertension will be the main focus of this review. Pulmonary arterial hypertension arises spontaneously, hereditarily, or as a complication of liver cirrhosis, connective tissue disease, HIV infection, congenital heart disease, schistosomiasis, or drug and toxin use (table 1).11 Symptoms of pulmonary arterial hypertension and other types of pulmonary hypertension are non-specific, frequently leading to delays in diagnosis and treatment.
The first instance of pulmonary hypertension was reported in 1891 by Ernst von Romberg, whereas Paul Wood was the first to describe the clinical and haemodynamic features of pulmonary hypertension in 1952.12 13 The First World Symposium on Pulmonary Hypertension convened in Geneva in 1973 and defined the disease, at that time known as primary pulmonary hypertension, clinically as a mean pulmonary artery pressure of >25 mm Hg at rest.14 This value was chosen rather arbitrarily because at that time, in healthy individuals, supine mean pulmonary artery pressure rarely exceeded 15 mm Hg at rest, was affected minimally by age, and almost never exceeded 20 mm Hg.14 This definition remained until the Sixth World Symposium on Pulmonary Hypertension in 2018, which redefined pulmonary hypertension as mean pulmonary artery pressure >20 mm Hg and added pulmonary vascular resistance ≥3 Woods units to the definition of all forms of precapillary pulmonary hypertension.1
More recently, the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology and the European Respiratory Society published guidelines further reducing the cut-off value for pulmonary vascular resistance to 2 Woods units, based on the available data on the upper limit of pulmonary vascular resistance in healthy individuals.15 Although these lower cut-off values better reflect the normal ranges of pulmonary haemodynamic variables, they have not yet resulted in new therapeutic recommendations, and the efficacy of treatment of pulmonary arterial hypertension in patients with a pulmonary vascular resistance of 2-3 Woods units or mean pulmonary artery pressure of 21-24 mm Hg is unknown. The new haemodynamic definitions in the 2022 guidelines of the European Society of Cardiology-European Respiratory Society have yet to be accepted by all major pulmonary hypertension research and clinical entities in the US and internationally, and therefore in this review, we will continue to refer to the definitions of the 2018 World Symposium on Pulmonary Hypertension.
The first medical treatment approved by the US Food and Drug Administration to treat pulmonary arterial hypertension was the synthetic prostacyclin, epoprostenol, in 1995.16–18 Progress was made in the following decades in identifying more causes of pulmonary arterial hypertension and establishing new diverse treatments. Currently, 14 drugs have been approved by the FDA. These drugs target vasodilatory pathways, including the endothelin, nitric oxide, and prostacyclin pathways.18 These treatments improve the symptoms and survival of patients with pulmonary arterial hypertension, but mortality remains high; the three year survival rate for patients with a high risk diagnosis is 28-55%.19–21 In patients who have unsatisfactory responses to treatment despite vasodilator treatment, lung transplantation might be necessary but is associated with its own inherent high risks of morbidity and mortality.22 Survival for patients with pulmonary hypertension associated with chronic lung disease has been reported to be even worse than for pulmonary arterial hypertension.23 24
This review provides an overview of the pathophysiology of pulmonary hypertension as it affects the lung and right ventricle. Most data on the pathophysiology of pulmonary hypertension come from models of pulmonary arterial hypertension and from biospecimens of patients with pulmonary arterial hypertension, and many concepts established in the pathophysiology of pulmonary arterial hypertension are also found in other types of pulmonary hypertension. Hence, in this review, we will focus on pulmonary arterial hypertension, but will discuss the intricacies of other types of pulmonary hypertension where relevant. We will then discuss the strengths and limitations of current therapeutic interventions, followed by emerging and promising new therapeutic strategies targeting new pathways. Finally, we will highlight current knowledge gaps in pulmonary arterial hypertension and pulmonary hypertension research.
Sources and selection criteria
Literature searches were performed from December 2021 to December 2022. PubMed and Medline databases were searched from database conception to December 2022. We searched PubMed and Medline for papers in the English language with the search words pulmonary hypertension, pulmonary arterial hypertension, right ventricle, pathogenesis, treatment, pulmonary vasodilators, inflammation, metabolism, angiogenesis, sex hormones, bone morphogenetic receptor protein 2, proliferation, remodelling, and tyrosine kinase. These search words were used in various combinations. We read the abstracts of relevant titles to confirm their relevance (impact of the paper on the specialty, journal impact factor, citation number, publication date), and the full papers were then extracted. References from extracted papers were checked for any further relevant papers.
General principles in pulmonary hypertension
The pulmonary circulation usually is of low resistance, and pulmonary blood pressure is about a 10th of systemic pressure.25 Pulmonary hypertension is diagnosed when mean pulmonary artery pressure is >20 mm Hg.11 The primary pathology in pulmonary arterial hypertension is a progressive and profound pulmonary vasculopathy characterised by vasoconstriction, vascular remodelling of all layers of the vessel wall, and in situ thrombosis. In the most advanced stage of the disease, patients have plexiform lesions, vascular alterations characterised by complex vascular formations originating from remodelled pulmonary arteries.26 27 In group 2 pulmonary hypertension, left heart disease leads to increased pulmonary venous pressure and, ultimately, pulmonary hypertension.28 29 Vascular remodelling occurs as a consequence of increased venous pressure and increased shear stress, but typically is less pronounced than in pulmonary arterial hypertension. Chronic obstructive or interstitial pulmonary disease are accompanied by destruction of the lung parenchyma and, consequently, reduction of alveolar-capillary density, resulting in group 3 pulmonary hypertension.30 31 Hypoxic pulmonary vasoconstriction, fibrotic processes, and inflammation also contribute to vascular remodelling in group 3 pulmonary hypertension.32 33
Group 4 pulmonary hypertension is caused by pulmonary emboli or pulmonary artery obstructions, and subsequent reductions in functional cross sectional area of the pulmonary vascular bed and increases in pulmonary vascular resistance.34 35 Overperfusion of non-obstructed vessels with subsequent remodelling also contributes to the development of pulmonary hypertension in this context.36 The pathophysiology of group 5 pulmonary hypertension is diverse and not fully understood, but frequently includes a combination of the processes listed above. Also, chronic haemolysis with subsequent depletion of nitric oxide stores is a contributor to pulmonary hypertension related to haemolytic anaemias in group 5 pulmonary hypertension.25
Hallmarks of pathophysiology of pulmonary arterial hypertension
The pathogenesis of pulmonary arterial hypertension is driven by the crosstalk between multiple cell types in the lung, including vascular cells, immune cells, and circulating cells. Pulmonary artery endothelial cells, pulmonary artery smooth muscle cells, and pulmonary artery fibroblasts are the major cells involved in the pathogenesis. Pulmonary artery endothelial cell dysfunction arises from several causes, such as shear stress, direct vascular injury, and intrinsic abnormalities.37 38 Endothelial dysfunction results in decreased production of vasodilating agents (eg, nitric oxide and prostacyclin) and increased synthesis of procontractile mediators (eg, endothelin 1).39 Also, increased production of growth factors and proinflammatory cytokines stimulate the proliferation of pulmonary artery smooth muscle cells and deposition and remodelling of the extracellular matrix.40
Abnormalities in the signalling pathways of transforming growth factor β and bone morphogenetic receptor protein 2 are key drivers of the development of pulmonary arterial hypertension (and, to some degree, also pulmonary hypertension). Bone morphogenetic receptor protein 2 is a major survival and homeostasis factor for pulmonary artery endothelial cells and pulmonary artery smooth muscle cells. Loss-of-function mutations in the bone morphogenetic receptor protein 2 gene (BMPR2) are responsible for heritable pulmonary arterial hypertension in most (>80%) patients.41 These mutations in BMPR2 promote abnormal proliferation of pulmonary artery endothelial cells and pulmonary artery smooth muscle cells. Decreased activation of the bone morphogenetic receptor protein 2 pathway has also been described in non-heritable types of pulmonary arterial hypertension.42 43 In contrast, increased activity of the transforming growth factor β pathway is a critical mediator of pulmonary artery endothelial cell and pulmonary artery smooth muscle cell dysfunction in pulmonary arterial hypertension.42 43 Many other molecular abnormalities have been described in pulmonary arterial hypertension and pulmonary hypertension but are beyond the scope of this review; these are reviewed elsewhere.18 42 43 In the following sections, we will highlight several key concepts important for the development and progression of pulmonary arterial hypertension and pulmonary hypertension.
Inflammation and immune dysregulation
Inflammation is a phenomenon commonly found in various types of pulmonary hypertension.44 A strong inflammatory response exists in pulmonary arterial hypertension associated with connective tissue disease, but substantial inflammation is also seen in idiopathic pulmonary arterial hypertension and hereditary pulmonary arterial hypertension. Robust immune responses are also seen in group 3 pulmonary hypertension and chronic thromboembolic pulmonary hypertension.45 46 Recently, a concept of immune phenotypes in pulmonary arterial hypertension groups has emerged.47 Pulmonary artery endothelial cells and inflammatory cells are critical local sources and targets of chemokines and cytokines, leading to pulmonary vascular remodelling in pulmonary hypertension. Interleukin 6 and interleukin 1β are prominent proinflammatory cytokines in pulmonary arterial hypertension and can directly control proliferation, migration, and differentiation of pulmonary artery endothelial cells, pulmonary artery smooth muscle cells, and immune cells.44 48–50 Autoantibodies and local lymph follicles have been implicated in promoting inflammation and immune activation in pulmonary arterial hypertension.19 20
BMPR2 mutations increase levels of proinflammatory cytokines (eg, interleukin 1β, interleukin 6).44 48 51 Similarly, a diminished bone morphogenetic receptor protein 2 signalling pathway in other forms of pulmonary arterial hypertension can lead to inappropriate expression of growth factors and proinflammatory responses in vascular cells, as described in experimental and human pulmonary arterial hypertension.44 51–53 Furthermore, BMPR2 loss of function in pulmonary artery endothelial cells causes a reduction in the vasoprotective peptide, apelin,54 as well as increased levels of fibroblast growth factor 2, mitogen activated protein kinases activity, interleukin 1β, and interleukin 6.48 49 55 Interleukin 1β and interleukin 6 can induce fibroblast growth factor 2 in pulmonary artery endothelial cells.50 Fibroblast growth factor 2 and interleukin 6 produced and released by pulmonary artery endothelial cells have an integral role in mediating proliferative responses of pulmonary artery smooth muscle cells and pulmonary artery fibroblasts.48 49 55
Patients with idiopathic pulmonary arterial hypertension show increased serum levels of chemokines that induce recruitment and migration of leucocytes.56–59 For example, C-C motif chemokine ligand 5 (CCL5), also called regulated on activation, normal T cell expressed and secreted (RANTES), exerts chemotactic power on monocytes and T cells.60–62 Furthermore, CCL5 can increase mitogenic activity through endothelin 1, thus inducing vasoconstriction.61 63 CCL2, also known as monocyte chemoattractant protein 1, is another chemokine involved in inflammation in pulmonary arterial hypertension.44 62 64 On stimulation by cytokines, such as interleukin 1β and interleukin 6, CCL2 is produced in several cell types, including monocytes, pulmonary artery endothelial cells, and pulmonary artery smooth muscle cells, participates in recruitment of monocytes to inflammation sites, and has a role in the proliferation and dedifferentiation of pulmonary artery smooth muscle cells. Serum levels of interleukin 1β, interleukin 6, and monocyte chemoattractant protein 1 are raised in patients with idiopathic pulmonary arterial hypertension.44 46 62 64
Patients with severe idiopathic pulmonary arterial hypertension show increased infiltration of the pulmonary vessel wall with macrophages,46 65 66 and T and B lymphocytes, suggesting a role for infiltration of immune cells in promoting vasoconstriction and lung vascular remodelling.44 62 67 68 Cytokines such as interleukin 1β, interleukin 6, and monocyte chemoattractant protein 1 cause exaggerated contractility and proliferation of vascular cells.48 Figure 1 provides an overview of the major inflammatory pathways involved in the pathogenesis of pulmonary arterial hypertension.
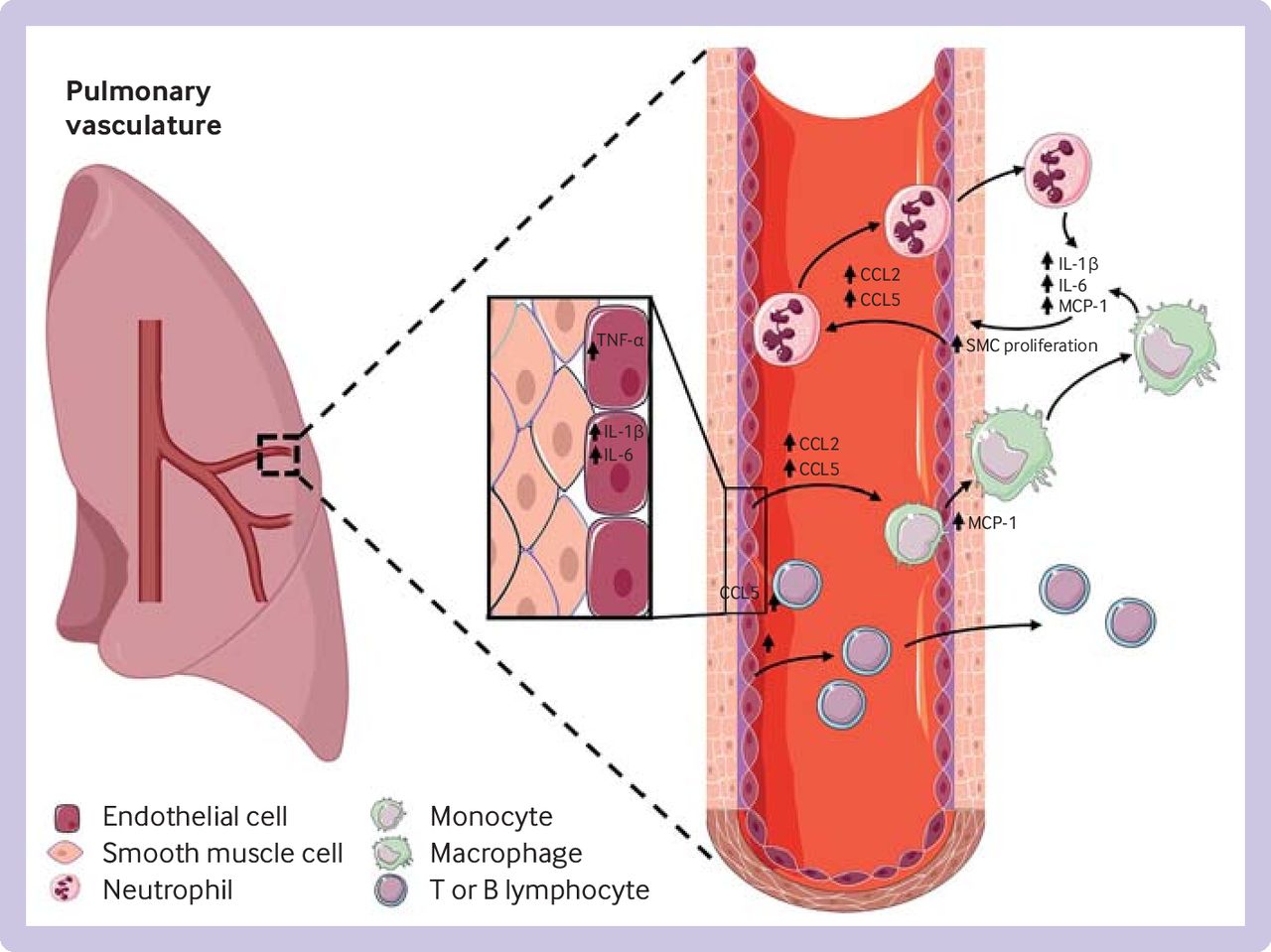
Overview of major inflammatory pathways involved in the pathogenesis of pulmonary arterial hypertension. Initial vascular injuries lead to increase in cytokines in vascular cells. After secretion into the bloodstream, cytokines promote accumulation of circulating neutrophils and monocytes, and T and B lymphocytes, resulting in increased vascular proliferation and damage. TNF-α=tumour necrosis factor α; IL=interleukin, MCP=monocyte chemoattractant protein, SMC=smooth muscle cell, CCL=C-C motif chemokine ligand
Based on the important role of inflammation in pulmonary arterial hypertension, several trials targeting components of the inflammatory cascade have been completed or are ongoing,69–71 but the results so far have not been consistent. For example, a recent trial of rituximab in patients with scleroderma-pulmonary arterial hypertension showed a non-significant increase in the six minute walk distance test.71 The mixed results of treatments targeting inflammation might indicate heterogeneity in the study populations or in the time course or extent of the inflammatory process.
Changes in angiogenesis
Dysfunctional angiogenesis is a major contributor to the development of pulmonary arterial hypertension. Blood vessels in the lung are lined by a pulmonary artery endothelial cell monolayer that has a crucial role in maintaining vascular homeostasis, and changes in pulmonary artery endothelial cells caused by shear stress, toxins, or genetic abnormalities lead to dysfunctional angiogenesis.72 Pulmonary artery endothelial cell dysfunction is a major characteristic of pulmonary arterial hypertension, and different altered pulmonary artery endothelial cell phenotypes are described during progression of pulmonary arterial hypertension (figure 2). During the early stages of vascular injury, pulmonary artery endothelial cells are proapoptotic, leading to a loss of pulmonary microvessels.73–76 Among the cells that survive the first insult, an endothelial cell apoptosis resistant phenotype is postulated to then emerge,77 78 associated with a hyperproliferative phenotype that contributes to formation of plexiform lesions and obliteration of lung vessels.78–81 Finally, in the terminal stages of pulmonary arterial hypertension, pulmonary artery endothelial cells might shift to a senescent phenotype, making the disease irreversible.82
Pulmonary artery endothelial cell dysfunction is associated with reduced pericyte coverage on pulmonary microvessels.83 This reduction of endothelial-pericyte interactions is explained by defects in pericyte motility and polarity, leading to progressive small vessel loss.84 85 Dysfunctional endothelial progenitor cells have been identified as contributors to angiogenesis dysfunction and development of pulmonary hypertension.86 Four different types of plexiform lesions have recently been identified, leading to a theory that bronchial artery anastomoses might contribute to the development of plexiform lesions.87

Current theory of change in endothelial cell phenotype in pulmonary artery hypertension and progression to irreversible disease. During the early stages of pulmonary artery hypertension, initial endothelial cell injury (shear stress, hypoxia, inflammation) leads to endothelial cell apoptosis (1), causing a reduction in pulmonary vessels. The remaining endothelial cells become hyperproliferative and resistant to apoptosis (2), leading to formation of plexiform lesions. In the terminal stages of pulmonary artery hypertension, endothelial cells become senescent (3), making the disease irreversible.
Because bone morphogenetic receptor protein 2 is crucial for pulmonary vascular homeostasis, impaired signalling of this protein is likely a major cause of dysfunctional angiogenesis in pulmonary arterial hypertension.88–90 The involvement of vascular endothelial growth factor in pulmonary arterial hypertension is also particularly important. Several studies have highlighted raised plasma levels of vascular endothelial growth factor in patients with severe pulmonary arterial hypertension.91–94 Also, expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 is strongly increased in pulmonary complex vascular lesions of patients with pulmonary arterial hypertension.90 95 Changes in other vascular homeostasis mediators, such as apelin, the angiopoietin system, platelet derived growth factor, nitric oxide, and others have also been described.54 96–98 Dysfunctional angiogenesis likely also contributes to the development of pulmonary hypertension in chronic thromboembolic pulmonary hypertension.99 Angiogenesis is currently not targeted directly by treatments for pulmonary arterial hypertension, although new treatments targeting bone morphogenetic receptor protein 2 or transforming growth factor β signalling (reviewed below) will likely have a major effect on the function and angiogenesis of pulmonary artery endothelial cells.
Perturbations in metabolism
Abnormal metabolic remodelling has emerged as a major driver of the pathogenesis of pulmonary arterial hypertension,100 and a key principle in pulmonary arterial hypertension relates to the shift from oxidative phosphorylation to glycolysis, known as the Warburg effect.100 101 Hallmarks of altered metabolism in pulmonary arterial hypertension include increased cytoplasmic glycolysis and glutaminolysis as well as impairments in mitochondrial biogenesis and fatty acid oxidation.102 103
Increased cytoplasmic glycolysis is implicated in the development and progression of pulmonary arterial hypertension.76 Multiple studies have identified upregulation of this pathway in pulmonary artery endothelial cells104–107 and pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension.106 108 These studies describe how glucose utilisation largely shifts towards lactate generation and away from the tricarboxylic acid cycle (figure 3). This metabolic shift leads to a reduction in the efficiency of generation of ATP by the mitochondrial tricarboxylic acid cycle. Among the molecular markers associated with this effect, an increase in expression of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) and lactate dehydrogenase B is found in lung sections and isolated pulmonary artery endothelial cells, and in human lung tissue.109–111 Upregulation of lactate production in pulmonary artery endothelial cells is described in vitro.102

Overview of major glycolytic and mitochondrial changes in pulmonary arterial hypertension. Glycolysis is upregulated in pulmonary arterial hypertension, and characterised by an increase in expression of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) and lactate dehydrogenase-B (LDHB) in lung cells. Upregulation of LDHB is associated with inhibition of pyruvate dehydrogenase (PDH) by pyruvate dehydrogenase kinase (PDK), leading to accumulation of lactate in cytoplasm and reduced mitochondrial biogenesis. Reduction in generation of ATP by tricarboxylic cycle (TCA) occurs in pulmonary vascular cells, associated with reduction in quantity of mitochondrial DNA (mtDNA) and increase in production of reactive oxygen species (ROS). Changes in fatty acid oxidation have not been fully elucidated. These abnormalities have been found in pulmonary artery endothelial cells, pulmonary artery smooth muscle cells, and in the right ventricle. G6P=glucose 6-phosphate; F6P=fructose 6-phosphate; F2,6P2=fructose 2,6-bisphosphate; F1,6P2=fructose 1,6-bisphosphate
As well as changes in glucose metabolism, reduced mitochondrial metabolism occurs in pulmonary arterial hypertension. Mitochondrial abundance is decreased in pulmonary artery endothelial cells and pulmonary artery smooth muscle cells,104 112–114 and reduced oxygen consumption and mitochondrial DNA are found in pulmonary artery endothelial cells.104 112 Also, expression, activity, or both, of mitochondrial complex I and IV are reduced in pulmonary artery endothelial cells104 and small pulmonary arteries113 114 from patients with pulmonary arterial hypertension. Increased reactive oxygen species production was found in pulmonary artery endothelial cells in vitro, associated with a reduction in antioxidants such as superoxide dismutase 1 and 2.105 112 Mitochondrial network fragmentation is seen in pulmonary artery smooth muscle cells, as well as a reduction in mitochondrial respiration.115 One of the central mechanisms involved in the Warburg effect is inhibition of pyruvate dehydrogenase.116 This enzyme has a crucial role in catalysing mitochondrial production of acetyl-coenzyme A from pyruvate, the end product of glycolysis. In pulmonary arterial hypertension, increased pyruvate dehydrogenase inhibitory kinase activity is seen in the pulmonary arteries of patients with pulmonary arterial hypertension,117–119 leading to uncoupled glycolysis and reduced mitochondrial pyruvate utilisation (figure 3).116
Impairments in fatty acid oxidation in pulmonary arterial hypertension are less well characterised. Whereas some studies described a reduction in fatty acid oxidation mediators in pulmonary artery endothelial cells in vitro (eg, 13C-α-ketoglutarate and acetyl-CoA acetyltransferase 2),106 112 others highlighted an increase in long and medium chain free fatty acid products, suggestive of lipotoxicity.102 109 Lastly, upregulation of glutamate metabolism is found in both the lung and right ventricle in patients with pulmonary arterial hypertension, and this finding has been linked to a proproliferative phenotype.109 120–122
Metabolic changes have also been described in other cell types. In fibroblasts from patients with idiopathic pulmonary arterial hypertension, an increase in aerobic glycolysis and in free reduced nicotinamide adenine dinucleotide (NADH) and NADH/nicotinamide adenine dinucleotide (NAD)+ ratios was seen.123 Immune cells in pulmonary arterial hypertension are highly glycolytic and the cytokines they produce affect mitochondrial function in the pulmonary vasculature.124 In the right ventricle, similar metabolic alterations are described and include an increase in aerobic glycolysis and a reduction in fatty acid oxidation and mitochondrial metabolism (see section on Abnormalities of right ventricle for details).
Clinically, patients with pulmonary arterial hypertension have substantial insulin resistance and dyslipidaemia.125 Some of these changes have been linked to worse outcomes.102 In a subset of patients with single nucleotide polymorphisms in genes encoding for sirtuin 3 and uncoupling protein 2, use of the pyruvate dehydrogenase kinase inhibitor, dichloroacetate, was associated with less severe pulmonary arterial hypertension.117
Epigenetics and genetics in pulmonary arterial hypertension
Major technological advances in genetic and epigenomic sequencing have rapidly increased our understanding of the contributions of these processes to the predisposition, pathobiology, and severity of pulmonary arterial hypertension.126 127 Exome sequencing and survey of patient cohorts have identified several mutations likely to have a causal role in pulmonary arterial hypertension, including BMPR2, EIF2AK4, TBX4, ATP13A3, GDF2, SOX17, AQP1, ACVRL1, SMAD9, ENG, KCNK3, and CAV1.126 128–140 Mutations in BMPR2 are the most commonly occurring and best characterised. BMPR2 mutations have been identified in 70-80% of patients with heritable pulmonary arterial hypertension and in 10-20% with idiopathic pulmonary arterial hypertension, and have been linked to increased susceptibility, earlier disease onset, and increased severity and mortality of the disease.126 141
Despite their proposed causal role in pulmonary arterial hypertension, these mutations are rare and have low penetrance, suggesting that additional modifiers or hits are needed to develop pulmonary arterial hypertension. Single nucleotide polymorphisms are genes that differ from normal genes by one nucleotide and can alter the function of the encoded protein. Although single nucleotide polymorphisms occur in a substantial proportion of the population, they could be important modifiers to explain susceptibility to pulmonary arterial hypertension or responsiveness to treatment.142 For example, single nucleotide polymorphisms in sirtuin 3 and uncoupling protein 2 play a part in drug metabolism, affecting the responsiveness of patients to treatment with dichloroacetate.117
Epigenetic modifiers influence how cells control gene expression and activity through processes such as DNA methylation, acetylation, histone modification, and RNA based modifications. DNA methylation at a gene promoter typically acts to repress gene transcription.143 Hypermethylation of SOD2 in patients with pulmonary arterial hypertension and in rodent models of pulmonary hypertension has been shown to contribute to proliferative antiapoptotic signalling in pulmonary artery smooth muscle cells.144 Global differences were also seen in DNA methylation signatures in pulmonary artery endothelial cells and pulmonary artery smooth muscle cells from patients with pulmonary hypertension compared with cells from control individuals.145 146 An increase in DNA methyltransferase 3β expression was found in the lungs of pulmonary hypertension patients.147 Histone acetylation or deacetylation, controlled by histone acetyltransferases and histone deacetylases, modifies the structure of histones by facilitating or inhibiting DNA access to transcriptional machinery. Results for inhibition of histone deacetylase in pulmonary arterial hypertension have been inconsistent, likely representing different contributions from various subtypes of histone deacetylase.148 149 Bromodomain containing protein 4, a member of the bromodomain and extra-terminal domain family that docks to acetylated histones, is upregulated in the lungs, distal pulmonary arteries, and pulmonary artery smooth muscle cells of patients with pulmonary arterial hypertension.150 Inhibition of bromodomain containing protein 4 is beneficial in experimental pulmonary arterial hypertension, and a clinical trial of inhibition of bromodomain containing protein 4 with apabetalone is currently ongoing (NCT04915300).151
Non-coding RNAs, like microRNAs (miRNAs), can epigenetically modify DNA to affect many biological processes implicated in the pathogenesis of pulmonary arterial hypertension.152–154 For example, changes in miR-424, miR-503, miR-204, and miR-143 signalling have been linked to changes in cell phenotypes and promotion of the pathogenesis of pulmonary arterial hypertension.127 Therapeutic strategies targeting miRNAs are limited by challenges in the pleiotropic effects of miRNAs, safety, and efficacy of delivery.155
Sex hormone signalling in pulmonary arterial hypertension
The prevalence of pulmonary arterial hypertension is higher in female individuals, with a female to male ratio in all forms of pulmonary arterial hypertension of 1.4-4.1 to 1.156 157 Despite the increased prevalence, female patients with pulmonary arterial hypertension have more favourable haemodynamic changes and improved clinical and survival outcomes.158–161 Furthermore, compared with male patients, female patients with pulmonary arterial hypertension have better right ventricular function, preserved right ventricular ejection fraction, better right ventricular-pulmonary artery coupling, and better right ventricular response to treatment.159–162 These more favourable haemodynamic variables are blunted in women after the menopause,158 and premature menopause is a major risk factor for the development of pulmonary hypertension.163 Sex hormones are modifiers of these phenomena. For example, superior right ventricular function in women correlates with plasma levels of 17β-oestradiol in health and pulmonary arterial hypertension.160 164 165 Conversely, other studies have linked oestrogens and their metabolites to the development of pulmonary arterial hypertension,166 167 and higher levels of oestrogen (from drug treatments or because of pregnancy) were associated with pulmonary arterial hypertension.168–170
Although most studies have focused on the role of 17β-oestradiol in pulmonary arterial hypertension, other sex hormones have been reported to have effects. For example, decreased levels of dehydroepiandrosterone were associated with the development of pulmonary arterial hypertension in men.171 In men, and in women after the menopause, lower levels of dehydroepiandrosterone and higher levels of 17β-oestradiol were associated with worse haemodynamic variables and right ventricular function and an increased risk of death.172 Another study found that low levels of dehydroepiandrosterone sulphate and high levels of testosterone correlated with worse right ventricular function in male patients with pulmonary arterial hypertension.173 Together, these data suggest that sex hormones have a vital role in the pathogenesis of pulmonary arterial hypertension and right ventricular adaptation.
17β-Oestradiol has nuanced cell-dependent and context-dependent and temporal effects on the pulmonary vasculature. For example, 17β-oestradiol increased proliferation of human pulmonary artery smooth muscle cells in some studies,167 174 175 whereas another study reported inhibitory effects of 17β-oestradiol on proliferation of human pulmonary artery smooth muscle cells and no effect on human pulmonary artery fibroblasts.176 Conversely, pulmonary artery endothelial cells from mice showed differences specific to sex in mitochondrial respiration, proliferation, and response to stress without the addition of exogenous sex hormones.177 Pulmonary artery endothelial cells derived from male individuals showed increased proliferation compared with cells derived from female individuals, and hypoxia or use of antimycin A induced an apoptotic response in pulmonary artery endothelial cells from female individuals but a necrotic response in cells from male individuals 178 In addition to the cell specific effects of sex hormones, other potentially confounding factors include whether the cells were from control individuals or patients with pulmonary arterial hypertension, and the location within the vascular tree where the cells were derived.
In contrast with the effect of 17β-oestradiol on the pulmonary vasculature, 17β-oestradiol is uniformly protective against right ventricular failure.179 180 We recently showed that 17β-oestradiol preserves and rescues right ventricular function and increases survival in rodent models of pulmonary arterial hypertension.54 The protective effects of 17β-oestradiol on the right venticle are mediated by oestrogen receptor α-mediated upregulation of bone morphogenetic receptor protein 2 and apelin in cardiomyocytes.54 These studies suggest that the effect of sex hormones might have differing roles in susceptibility versus survival and right ventricular adaptation in pulmonary arterial hypertension. Three clinical trials focusing on sex hormone signalling are currently ongoing (see section on Current treatments).
Abnormalities of right ventricle
Right ventricular function predicts survival in patients with all forms of pulmonary hypertension,20 181–184 but the pathophysiology of right ventricular failure is poorly understood. Research has identified changes in inflammatory mediators, neutrophil and macrophage infiltration, microvascular dysfunction, apoptosis, oxidative stress, metabolic shifts, fibrosis, and mechanical alterations as mediators of right ventricular failure.184–189 Figure 4 summarises changes in the right ventricle in pulmonary arterial hypertension.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Changes in the right ventricle in pulmonary hypertension. In early pulmonary hypertension, the right ventricle undergoes compensatory changes, including angiogenesis, hypertrophy of cardiac myocytes, and increased wall thickness, and right ventricle and pulmonary artery afterload remain coupled. In late pulmonary hypertension, the right ventricle undergoes maladaptive changes, including metabolic reprogramming, capillary rarefaction, fibrosis, apoptosis, inflammation and immune cell recruitment, oxidative stress, and neurohormonal and dysregulation of the renin-angiotensin-aldosterone system (RAAS); the right ventricle dilates and the septum bows into the left ventricle, and right ventricle and pulmonary artery load become uncoupled. Original figure created with BioRender.com. ROS=reactive oxygen species
The first phase of right ventricular adaptation to pulmonary arterial hypertension or pulmonary hypertension is characterised by compensatory right ventricular hypertrophy and an increase in contractility of 4-5-fold in response to its increased load.183 190 If severe pulmonary hypertension persists or worsens, the right ventricle enters a maladaptive phase in which rising wall stress, right ventricular dilation, and right ventricle-pulmonary artery uncoupling occur, progressing to right ventricular failure.183 191 192 What factors predict or result in right ventricular failure in some patients but not in others is not known.
Clinical manifestations of right ventricular failure include dyspnoea and exercise limitation in early stages, followed by retention of fluids, arrhythmias, syncope, and organ failure, including kidney failure and congestive hepatopathy.193–197 Because of the interdependence of the right and left ventricles, dyssynchronous contraction and right ventricular dilation causes bowing of the septum into the left ventricle, resulting in impaired left ventricular compliance and diastolic filling.198–200 In patients presenting with acute right ventricular failure, mortality is estimated at 40% in those requiring hospital admission,201–204 with some patients presenting with sudden cardiac death.205
Insufficient angiogenesis is thought to be a factor in right ventricular maladaptation.206 One theory is that vascular rarefaction results in decreased delivery of oxygen or metabolic substrates.188 Some studies indicated a potential capillary drop-out in the right ventricle in experimental pulmonary hypertension.120 207 Studies that used stereology techniques, however, found increased compensatory angiogenesis correlating with increased cardiomyocyte volume and an increase in the radius of tissue served per vessel in the right ventricle in human end stage pulmonary arterial hypertension191 as well as in rodent models of pulmonary hypertension.208 209 In one of these studies, arterial delivery of metabolic substrates was decreased but evidence that this effect was functionally significant did not exist, because no tissue hypoxia or depletion of key metabolic substrates was found.208 Differences in experimental techniques and the clinical characteristics of tissues might explain this discrepancy. More studies are needed to determine the role of angiogenesis in right ventricular (mal)adaptation.
In healthy adults, the primary energy source is fatty acids, accounting for 60-90% of the substrate used for generation of ATP.210 211 In experimental pulmonary hypertension, right ventricular metabolic reprogramming leads to substantial shifts in substrate utilisation, with increased uptake of glucose by the right ventricle and anaerobic glycolysis. This effect has been shown with 2-[18F] fluoro-2-deoxy-D-glucose positron emission tomography in the lung and right ventricle of patients with pulmonary arterial hypertension.212 Concomitantly, decreased fatty acid oxidation in the right ventricle is seen, resulting in a return to a fetal-like metabolic state.208 213–215 This dysregulated fatty acid oxidation leads to lipid accumulation and production of toxic intermediates, such as ceramide or palmitate.13–17 Also, this switch towards glucose provides signals and building blocks for structural remodelling of the heart.216 Because this process cannot be explained by substrate deprivation, metabolic reprogramming might result from intrinsic changes within the myocytes of the right ventricle.208 Several trials focusing on modifying metabolism in pulmonary arterial hypertension have been completed or are currently ongoing. In a pilot trial, use of metformin was associated with better right ventricular function and less lipid accumulation.217
Right ventricular fibrosis is known to occur in pulmonary arterial hypertension and pulmonary hypertension, particularly at right ventricular septal insertion points, and is thought to be a major component leading to right ventricular failure.218–220 New imaging methods with cardiovascular magnetic resonance have quantified myocardial extracellular volume, a histologically validated marker of diffuse interstitial fibrosis.221–223 Extracellular volume measured by cardiovascular magnetic resonance correlates with known echocardiographic prognostic markers of right ventricular systolic function.224 225 Despite substantial progress in this field, more research is needed into the biology and pathophysiology of the right ventricle.226
Other pathophysiological features
As well as the hallmarks of the pathogenesis of pulmonary arterial hypertension, several other features occur in the lung, right ventricle, or both, in pulmonary arterial hypertension, such as endothelial-to-mesenchymal transition, vascular fibrosis, and perturbation of signalling hubs. Mechanisms not dependent on transforming growth factor β, driven by oxidative stress (eg, neural precursor cell expressed developmentally downregulated protein 9, NEDD9) have recently been found to promote endothelial fibrosis and pulmonary artery remodelling. These mechanisms are not described here, but are highlighted in recent articles.227–229
Current treatments
The currently available medical treatments for pulmonary arterial hypertension target the three main pathways involved in the control of pulmonary vascular tone and proliferation: prostacyclin pathway, with prostacyclin analogues and prostacyclin receptor agonists (epoprostenol, treprostinil, iloprost, selexipag) that induce vasodilation by increasing levels of cyclic AMP; nitric oxide pathway, targeted by phosphodiesterase inhibitors (sildenafil, tadalafil) and by the guanylate cyclase activator, riociguat; and endothelin pathway, targeted by receptor antagonists (bosentan, ambrisentan, macitentan). These drug treatments, or a combination of treatments, such as tadalafil and ambrisentan in the AMBITION (A Study of First-Line Ambrisentan and Tadalafil Combination Therapy in Subjects With Pulmonary Arterial Hypertension) trial,230 are the mainstay of treatment of pulmonary arterial hypertension, and have been shown to benefit patients through improvements in exercise capacity, quality of life, and time to clinical worsening.231–241 Survival benefit has only been found with epoprostenol,16 although meta-analyses and post hoc survival analyses have suggested survival benefit from upfront combination dual or triple treatment.242–244 Riociguat is the only approved medical treatment for group 4 pulmonary hypertension, and has been shown to improve exercise capacity and haemodynamic variables in patients with inoperable or recurrent chronic thromboembolic pulmonary hypertension.236
Pulmonary artery thromboendarterectomy is the treatment of choice for chronic thromboembolic pulmonary hypertension.245 Balloon pulmonary angioplasty can be used in inoperable cases. Recently, inhaled treprostinil became the only treatment approved by the FDA for patients with group 3 pulmonary hypertension after the drug was shown to improve exercise capacity in patients with pulmonary hypertension caused by interstitial lung disease.246 Recent guidelines and reviews provide a detailed overview of the treatment strategies for pulmonary hypertension.15 247 248
Emerging therapeutic directions
Current treatments for pulmonary arterial hypertension are efficacious but are limited by their inability to reverse pulmonary vascular remodelling. Several interventions targeting new pathways involved in the pathogenesis of pulmonary arterial hypertension are currently in clinical trials, with the aim of establishing curative treatments. A detailed review has recently been published.249
Sex hormone signalling
Sex hormones have a dynamic and intrinsic role in the cardiopulmonary unit, regulating tissue homeostasis, response to injury, and pathogenesis of the disease. Treatments designed to target the synthesis, metabolism, or receptor signalling pathways of sex hormones, therefore, represent new approaches to treat patients with pulmonary arterial hypertension. The EDIPHY (Effects of Dehydroepiandrosterone in Pulmonary Hypertension) trial, an ongoing crossover phase 2 clinical trial (NCT03648385), explores whether administration of dehydroepiandrosterone for 18 weeks affects right ventricular function in patients.250 Dehydroepiandrosterone is a metabolic precursor of testosterone and oestradiol, and is decreased in pulmonary arterial hypertension.171 PHANTOM (Pulmonary Hypertension and Anastrozole Trial), a phase 2 trial, determined whether inhibiting aromatase and reducing oestradiol levels with anastrozole improved the primary endpoint of the six minute walk distance test in pulmonary arterial hypertension (NCT03229499).251 Finally, T3PAH (Tamoxifen Therapy to Treat Pulmonary Arterial Hypertension) is a phase 2 trial, determining whether the selective oestrogen receptor modulator, tamoxifen, changes right ventricular tricuspid annular plane systolic excursion (NCT03528902). The effects of tamoxifen on the activity of the oestrogen receptor in the right ventricle have not been evaluated, however, and tamoxifen can act as an oestrogen receptor agonist in non-gonadal tissues (eg, bone).252 253 Preclinical studies showed that 17β-oestradiol and oestrogen receptor α are protective against right ventricular failure induced by pulmonary arterial hypertension. The effects of anastrazole and tamoxifen on right ventricular function are therefore being closely monitored.
Stimulating bone morphogenetic receptor protein 2 signalling
As previously discussed, disruption of the bone morphogenetic receptor protein 2 pathway is implicated in the pathogenesis of pulmonary arterial hypertension, and BMPR2 mutations are the most commonly identified form of heritable pulmonary arterial hypertension. Studies have shown that conditional deletion of BMPR2 in endothelial cells is sufficient to induce spontaneous pulmonary hypertension in mice,254 and that rescue of endothelial bone morphogenetic receptor protein 2 expression reverses experimental pulmonary hypertension.255 256 In preclinical trials, FK506 (tacrolimus) increased bone morphogenetic receptor protein 2 signalling.257 FK506 also reduced right ventricular fibrosis, stabilised right ventricular capillarisation, and improved right ventricular function in experimental pulmonary hypertension independent of its beneficial effects on pulmonary vascular remodelling.258 A phase 2 trial of FK506 showed that low doses of FK506 were well tolerated in patients with pulmonary arterial hypertension with improvements in walk distance and in biomarkers for right ventricular function.259 However, phase 2b or phase 3 trials are not currently ongoing.
Recent studies highlighted the potential role of sotatercept, a fusion protein that acts as a ligand trap for transforming growth factor β members and that restores the balance between growth promoting and growth inhibiting bone morphogenetic protein pathways.260 A multicentre, randomised, double blind, phase 2 trial of sotatercept for background treatment in patients with pulmonary arterial hypertension showed an improvement in exercise capacity and reduction in pulmonary vascular resistance at 24 weeks.260 Several phase 3 trials of sotatercept are underway or completed (NCT04576988, NCT04811092, and NCT04896008), with the results of the STELLAR (A Study of Sotatercept for the Treatment of Pulmonary Arterial Hypertension) trial showing beneficial effects on exercise capacity and a reduction in pulmonary vascular resistance, resulting in a breakthrough therapy designation by the FDA, and a priority medicines designation by the European Medicines Agency.261
Rare mutations in GDF2, the gene encoding bone morphogenetic protein type 9, have been identified in patients with heritable pulmonary arterial hypertension.262 Reduced activity and plasma levels of bone morphogenetic protein types 9 and 10, the main ligands of activin receptor-like kinase 1-bone morphogenetic receptor protein 2 heterocomplexes, were found to be reduced in some patients with pulmonary arterial hypertension, including those not carrying GDF2 mutations.139 In some studies, bone morphogenetic protein 9−/− or wild-type mice given anti-bone morphogenetic protein 9 antibodies were protected from pulmonary hypertension, and inhibition of bone morphogenetic proteins 9 and 10 with a ligand trap in rats caused regression of pulmonary hypertension.263 However, other studies showed the contradictory finding that administration of bone morphogenetic protein 9 prevented and reversed established pulmonary hypertension in transgenic mice with spontaneous pulmonary hypertension caused by a mutation in the bone morphogenetic receptor protein 2 locus, as well as in rat models of pulmonary arterial hypertension.75 Together, these studies suggest that bone morphogenetic proteins 9 and 10 are possible new therapeutic targets in pulmonary hypertension, but the precise role of dysfunctional bone morphogenetic protein signalling and balance of bone morphogenetic protein ligands with the bone morphogenetic receptor protein 2 receptor needs to be further clarified.
Tyrosine kinase inhibition
The cancer hypothesis of pulmonary arterial hypertension, suggesting that pulmonary arterial hypertension and cancer share common features, such as hyperproliferation, apoptosis resistance, and metabolic reprogramming, has led to the exploration of repurposing treatments already tested in the treatment of malignancies.249 Among these, tyrosine kinase inhibitors, already approved and in use as antineoplastic agents in numerous types of cancer, are postulated to be beneficial in the treatment of pulmonary arterial hypertension by inhibiting kinases related to cell growth and suppressing pulmonary vascular angioprolilferative remodelling, and have been beneficial in preventing or reversing experimental pulmonary arterial hypertension.264–266
A randomised controlled trial of imatinib in pulmonary arterial hypertension showed improved exercise capacity and haemodynamic variables, but serious adverse events included several subdural haematomas, and discontinuation of the study drug was common.267 In contrast, a pilot trial of sorafenib in pulmonary arterial hypertension found improvement in right ventricular ejection fraction and exercise tolerance in some patients, but a concerning decrease in median cardiac index.268 A phase 2 trial of nilotinib (AMN107) was stopped early because of serious adverse events and was not powered for efficacy. Differences in the effects of tyrosine kinase inhibitors are likely because of the different tyrosine kinases being targeted. Several trials with oral or inhaled tyrosine kinase inhibitors are currently ongoing, including phase 2 trials of imatinib (NCT04416750) and seralutinib (NCT04456998).269 ,270 Seralutinib (targeting platelet derived growth factor receptor, colony stimulating factor 1 receptor, and c-KIT) was recently announced in a press release to have met the phase 2 primary endpoint of a decrease in pulmonary vascular resistance, with publication still pending.271
Targeting metabolism
Another main target is cellular metabolism. One strategy to reverse metabolic remodelling in pulmonary arterial hypertension is to target glycolytic inhibitors. Dichloroacetate, a pyruvate dehydrogenase kinase inhibitor, has shown promise by reducing mean pulmonary artery pressure and pulmonary vascular resistance and improving walking capacity in patients with pulmonary arterial hypertension.102 110 117 144 A pilot study showed improvement in haemodynamic variables in genetically susceptible patients.117
Improving fatty acid oxidation is another strategy to reduce pulmonary arterial hypertension. Metformin, an antihyperglycaemic agent, reduces lipid deposition in the right ventricle of BMPR2 mutant transgenic mouse models.272 Recently, a phase 2 clinical trial in patients with pulmonary arterial hypertension showed a statistically significant improvement in right ventricular fractional area and a reduction in right ventricular triglyceride content.217 Finally, treatments aimed at improving mitochondrial function have shown promising results. Supplementation with coenzyme Q improved mitochondrial respiration and echocardiographic markers of right ventricular function in individuals with pulmonary arterial hypertension.273 Pioglitazone, a peroxisome proliferator activated receptor γ agonist, restored mitochondrial function, reversed pulmonary arterial hypertension, and prevented right ventricular failure in rat models.274
In summary, several potentially effective new drug treatment strategies for pulmonary arterial hypertension are being explored, with several of these treatments already being tested in clinical trials. In particular, sotatercept has generated robust phase 2 data and a press release of phase 3 results.260 261 Also, non-drug treatments, such as pulmonary artery denervation, are currently being studied and seem promising.275
Questions for future research
Despite improvements in our understanding of pulmonary arterial hypertension, several questions remain. The development of curative treatments for pulmonary arterial hypertension and for other types of pulmonary hypertension is needed. Furthermore, the key initiating events in pulmonary arterial hypertension and pulmonary hypertension and in what combination they occur are still unclear. Also unclear is whether pulmonary arterial hypertension is a systemic rather than a cardiopulmonary disease, and whether pre-existing metabolic, inflammatory, and genetic and epigenetic abnormalities can predispose some individuals to develop pulmonary arterial hypertension or pulmonary hypertension. Regardless, the critical point at which a patientdevelops or does not develop pulmonary arterial hypertension or pulmonary hypertension is not defined. Molecularly, which cell types are the most critically injured is still debated, and the exact mechanisms are speculative because most work on the cause of pulmonary arterial hypertension and pulmonary hypertension has been in preclinical animal models (which do not replicate human disease completely), and patient tissues from early stages of the disease are not available.
Finally, despite its importance for patient survival and mortality, no treatments directed at the right ventricle currently exist. Research is only beginning to molecularly define right ventricular failure and how it progresses. The development of powerful new omics based approaches and improved access and affordability of database repositories increases the likelihood that we will identify new cell populations in the pulmonary circulation that are critical for the development of pulmonary arterial hypertension and pulmonary hypertension, define new signalling networks, and more accurately determine intercellular and intersystem communication in the pathogenesis of pulmonary arterial hypertension and pulmonary hypertension. These advances will ultimately lead to the development of new molecular targets and prognostic biomarkers. Algorithms based on machine learning will also help identify prognostic factors for pulmonary arterial hypertension and pulmonary hypertension. This approach will allow investigators to rapidly process large amounts of data, and despite the heterogeneity of the disease, use an unbiased approach to identify new patterns and indicators of the prognosis, progression, and outcomes of pulmonary hypertension. Clinically, more research is needed to clarify whether treating people according to new definitions means better outcomes. Also, the role and place of new treatments needs to be defined. Table 2 summaries the current gaps in our knowledge.
Selected current gaps in knowledge of pulmonary arterial hypertension
Conclusions
Over the past two decades, substantial progress has been made in understanding the molecular basis and clinical characteristics of pulmonary arterial hypertension and pulmonary hypertension. Pathophysiologically relevant and clinically targetable changes exist in the areas of inflammation and immune dysregulation, angiogenesis, metabolism, epigenetics and genetics, as well as sex hormone signalling. Major progress has also been made in identifying mediators of right ventricular adaptation and maladaptation in pulmonary hypertension. New treatment strategies focusing on these areas are promising and could change how we think about treatment in the near future.
Questions for future research
What molecular mechanisms initiate pulmonary arterial hypertension and which cell types are critical targets?
Can we establish curative treatments for pulmonary arterial hypertension and other types of pulmonary hypertension?
Is there a role for personalised treatment approaches in pulmonary arterial hypertension and other types of pulmonary hypertension?
Is there a role for pulmonary vasodilators for patients that do not meet enrolment criteria for previous trials?
References
Footnotes
Twitter @LabLahm
Contributors SB, RSF, and SG wrote the manuscript. AF and TL wrote and revised the manuscript. TL is the guarantor.
Funding Funding and support were provided by the American Heart Association Career Development Award 19CDA34660173 (AF), Bayer PHAB Level 3 Award (AF), NIH 1R01HL164791-01 (AF), VA Merit Review Award 2 I01 BX002042-05 (TL), NIH 1R01HL144727-01A1 (TL), NHLBI 1P01 HL158507-01 (TL), and Borstein Family Foundation, and Bayer Crosswalk PHAB_3.0 (RSF).
Competing interests We have read and understood the BMJ policy on declaration of interests and declare the following interests: none.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Commissioned; externally peer reviewed.